
Calculations performed between numeric protocol readouts, chemical properties, and constant numbers, can be defined in CDD Vault when the CDD Visualization extension is enabled in the Vault. Contact support to find out how to become a CDD Visualization subscriber.
In this article, we are looking at performing calculations between readouts of a single protocol and calculations with chemical properties. Go to this article for calculations that involve multiple protocols.
Basic steps to add a calculated readout
Readout definitions
Chemical propertiesAverage
Geomean
Median
Sum
Aggregation ScopeCalculations with protocol condition dependencies
pIC50
Geometric mean of IC50
Absolute IC50
FP ratio
BEI
LiPE (LLE)
Reagent Inventory
Calculations are based on existing assay data within your CDD Vault protocols, as well as chemical properties of the registered compounds, and numeric constants. In this manner, you can build dynamic mathematical functions that use protocol readouts as variables, and are automatically updated when new raw data is imported into the Vault.
Calculations are defined as new readouts in existing protocols, based on previously defined readouts. In a new protocol, calculations will be added after the initial set of raw data readout definitions.
We assume here that you are familiar with defining protocols and readouts already, and if you are new to protocol creation, please start here.
Basic steps to add a calculated readout
On the "Protocol Details" tab, and click "Add a readout definition".
In the readout definition pop up, set the Data Type to 'Calculated':
You will need to provide a name for the readout definition (that does not conflict with any other readout names in this protocol of course), and then fill in the formula in the provided formula builder.
Begin typing in the Formula box to see what kinds of functions can be applied and combined.
Type a left square bracket [ to bring up a list of available readout definitions.
Type a curly bracket { to reveal a list of chemical properties.
Auto-complete and syntax help will appear as you type.
Use parentheses ( ) to indicate the order of operations.
Reference Variables
Calculations performed on referenced variables are dynamic: when the underlying variables are changed, the calculation is updated automatically. In this way, a single calculation can be defined to be performed on previously imported data, and on any incoming data.
Readout definitions
For your calculation, you can reference other existing readout definitions from the current protocol. Make sure to define all of the basic readout definitions before you start to combine them using calculations. For example, if you plan to build a calculation that includes % Inhibition values, % Inhibition needs to be defined ahead of the calculation- otherwise it will not be available as a reference variable when you start to build your formula.
Syntax
Type in an open square bracket, then click a readout name in the drop-down list: the matching bracket will be inserted automatically. The list of available options includes all available readout definitions with your current protocol appearing first, followed by all other protocols in alphabetical order.
[Protocol name -> readout definition name (units)] e.g. [Primary Screen -> Raw Fluorescence (RFU)]
Scope
By default, when using above syntax, calculations are performed for each molecule/batch with data in the referenced readout in per readout row mode.
- If the referenced readout is blank for a given batch, no calculated value will be returned for this batch.
- If there are multiple values in the referenced readout for a given batch (replicate data), then multiple values will be returned by the calculation.
- If the calculation references two readout definitions, a calculated value will be returned only if both of the referenced readout values occur on the same row. This happens when the data are added to the protocol in a single import event, and are both on the same row of the import file.
Here's an example of taking the ratio of two readouts for a single batch: the ratio value is calculated only when both CFP (477 nm) and YFP (514 nm) values appear on the same row, even though there are individual data points for both readouts on two other rows.

To perform a calculation based on multiple readout rows per batch, such as the scenario shown in the screen-shot above, the referenced readout(s) should aggregated with Average or Median functions.
Chemical properties
When you import molecule structures into CDD Vault, chemical properties are automatically calculated, and displayed on the molecule details page, or in the search results. These properties may be referenced as variables within calculated readout definitions.
Syntax
Type in an open curly bracket, then click a chemical property name in the drop-down list: the matching bracket will be inserted automatically.
{chemical property name} e.g. {Molecular Weight}
Here's the list of available chemical properties:
- Molecular weight (g/mol)
- log P
- H-bond donors
- H-bond acceptors
- Lipinski violations
- logD
- logS (solubility model)
- pKa
- CNS MPO score
- Topological polar surface area
- Exact mass (g/mol)
- Fsp3
- Heavy atom count
- Rotatable bonds
- Formula weight (g/mol)
Scope
When a chemical property is referenced in a calculated readout, molecule level properties are used, with the exception of formula weight, which is calculated per batch of molecule.
Functions
The following functions can be performed on referenced readout definitions. These functions may be used as part of another calculation, and in fact should be used on replicate data from multiple runs, or imported from multiple files.
Average
Average returns an average of all readout values in the selected aggregation scope for the chosen readout definition. When performed as a standalone calculation (not part of a larger formula), the returned result also includes the standard deviation, and the number (n) of values.
output: Avg ± StDev (n)
23.43 ± 3.16 (n=8)
A bounded value (that contains "less than" or "greater than" numeric modifier) will not be included when computing the average if there is another measurement that is consistent with it and is more specific. Exact values are always included.
- Less specific bounded values are not included:
Average(<1, <10) = <1
Average(20, 30, <100) = 25
Average(80, <100, 150) = 115 (<100 is consistent with 80 and less specific) - Bounded values are retained if they are not consistent with an exact value:
Average(<2, 8) = <5
This rule is based on how drug discovery data is normally generated. For example, if you perform a concentration-response assay between 100 to 10,000 nM, and the IC50 comes out as <100 nM, you will test at lower concentrations, say between 1 and 200 nM. If the IC50 in the second test was 80 nM, it makes sense to leave out the <100 nM from the first assay, because it has been superseded by the more specific 80 nM measurement.
On the other hand, if you ran the 100 to 10,000 nM assay twice and the only IC50s you had were <100 and 120 nM, we shouldn’t ignore the <100 nM, because doing so would result in a number that was artificially high.
Geomean
Geomean returns a geometric average of all readout values in the selected aggregation scope of the chosen readout definition. When performed as a standalone calculation (not part of a larger formula), the returned result also includes the standard deviation, and the number (n) of values.
Geomean is automatically inserted when averaging CDD-computed EC50 values. To over-ride the geomean aggregation, use "mean" in the formula.
output: Avg ×/÷ StDev (n)
23.43 ×/÷ 3.16 (n=8)
A bounded value (that contains "less than" or "greater than" numeric modifier) will not be included when computing the average if there is another measurement that is consistent with it and is more specific. Exact values are always included.
- Less specific bounded values are not included:
Average(<1, <10) = <1
Average(20, 30, <100) = 25
Average(80, <100, 150) = 115 (<100 is consistent with 80 and less specific) - Bounded values are retained if they are not consistent with an exact value:
Average(<2, 8) = <5
This rule is based on how drug discovery data is normally generated. For example, if you perform a concentration-response assay between 100 to 10,000 nM, and the IC50 comes out as <100 nM, you will test at lower concentrations, say between 1 and 200 nM. If the IC50 in the second test was 80 nM, it makes sense to leave out the <100 nM from the first assay, because it has been superseded by the more specific 80 nM measurement.
On the other hand, if you ran the 100 to 10,000 nM assay twice and the only IC50s you had were <100 and 120 nM, we shouldn’t ignore the <100 nM, because doing so would result in a number that was artificially high.
Median
Median returns the median of all readout values in the selected aggregation scope for the chosen readout definition. When performed as a standalone calculation (not part of a larger formula), the returned result also includes the standard deviation, and the number (n) of values.
output: Median (n)
23.43 (n=8)
Sum
Sum function adds all readout values in the selected aggregation scope of the chosen readout definition. When performed as a standalone calculation (not part of a larger formula), the returned result also includes the number (n) of values.
output: Sum (n)
187.04 (n=8)
Syntax
average([Protocol name -> readout definition name (units)]) e.g. [Primary Screen -> Inhibition (%)]
geomean([Protocol name -> readout definition name (units) (CDD)]) e.g. geomean([Secondary Screen ->IC50 (uM)(CDD)])
sum([Protocol name -> readout definition name (units)]) e.g. sum([Inventory -> Amount Debited (mg)])
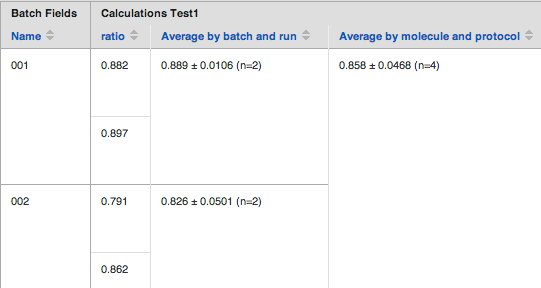
Aggregation Scope
Aggregation scope appears as a drop-down below the formula field. The same aggregation scopes apply to both average and median calculations.
- Aggregate by batch and run - A single aggregate value is calculated per batch of molecule, for each individual run of the current protocol. This is the best option for replicate data.
- Aggregate by molecule and protocol -A single aggregate value is calculated across all batches of a molecule and across all runs of the current protocol. Only one average value per molecule will ever be reported for the current protocol. This is the best summary level result.
- Aggregate by batch and protocol - An individual aggregate value is calculated for each batch of a molecule, across all runs of the current protocol. There will be as many average values, as there were tested batches of a molecule in a protocol. This is the best option to keep track of compound performance.
Here's an illustration of the different aggregation scope results:
Operators
Mathematical operations can be performed on constant numbers, readouts, including calculated readouts and chemical properties using the following operators
| * | multiplication |
| / | division |
| + | addition |
| - | subtraction |
| ^ | power : base^exponent |
| log() | logarithm base 10 : log(number) |
| ln() | natural logarithm, base e : ln(number) |
| exp() | e to the power of number : exp(number) |
Calculations using conditions
This is a more advanced topic - please be sure to have read the following two articles to understand protocol conditions and calculation syntax: article on cross protocol calculations as well as setting up conditions.
In brief - if any of your readouts are dependent on a condition, you may choose to integrate the condition into a calculated readout. Please note, that the more conditions you have defined in your protocol, the more complex the formulas may be become. This can become of importance if you change conditions well after protocol creation and having imported data to this protocol. Nevertheless, referencing to a condition within a calculated readout is of advantage for cross-protocol calculations.
As long as you are calculating only within a single protocol without cross-referencing, you don't necessarily have to specify the condition in the calculations. This has the additional advantage that you only require one calculated readout, instead of two (one for each condition). Though by all means you may want to define two separate columns nevertheless - for sake of clarity and, more importantly for the possibility of using them in cross-protocol calculations.
For example: A "Cytotox Assay" having two conditions (Hela, 3D7) may use this simple formula:
geomean([Cytotox Assay -> IC50 (uM) (CDD)])
In the search results table this results in two columns for the mean IC50, one for each condition, just as if you had defined these two separate calculations:
geomean([Cytotox Assay -> IC50 (uM) (CDD)]<[Cells]="HeLa">)
geomean([Cytotox Assay -> IC50 (uM) (CDD)]<[Cells]="3D7">)
Example calculations
pIC50
(calculation base on uM values, hence the additional factor 10E-6).
-log([Secondary Screen -> IC50 (uM)(CDD)] * 10^-6)
Geometric mean of the IC50
If you don't use pIC50s (you really should), then IC50 values can be averaged using the geometric mean, since they are based on a logarithmic scale of concentrations. When you average the CDD-calculated IC50, the formula will automatically default to the geometric mean.
geomean([Secondary Screen -> IC50 (uM)(CDD)])
output: Avg ×/÷ StDev (n)
23.43 ×/÷ 3.16 (n=8)
Absolute IC50
Use the calculated fit parameters from the Dose-Response calculation, and substitute the absolute inhibition value. We show the equation for 50% activity, but a similar calculation can be done at any other activity. Thus, the formula will be applicable to other IC## or EC## calculations as well.
= [Protocol Name-> EC50 (uM)]/((([Protocol Name-> Maximum Response (%)] - [Protocol Name-> Baseline Response (%)])/(50 - [Protocol Name-> Baseline Response (%)])-1)^(1/[Protocol Name-> Hill Slope]))
Fluorescence polarization ratio
([Primary Screen -> nFparallel]-[Primary Screen -> Fperpendicular])/[Primary Screen -> Fparallel]+[Primary Screen -> Fperpendicular]
Binding Efficiency Index
BEI is defined as (pIC50/molecular weight (kDa)). Based on the calculated IC50 from a protocol and molecular weight, reported in CDD in g/mol. Don't forget to convert IC50 to Molar units, and to convert molecular weight to KiloDaltons.
IC50(uM), MW (g/mol)
Create a pIC50 readout definition. Use the pIC50 to calculate BEI:
average([Secondary Screen -> pIC50]) / ({Molecular Weight} * 10^-3)
Lipophilic Efficiency
Lipophilic Efficiency is defined as (pIC50-cLogP). You will find clogP as part of the calculated properties: The logarithm of the partition coefficient. The implementation uses the approach described in Gedeck et al. (DOI: 10.1021/acs.jcim.7b00315) using publicly available log P data. (See more.)
Create a pIC50 readout definition. Use the pIC50 to calculate LiPE:
average([Secondary Screen -> pIC50]) - {log P}
Reagent Inventory
You can use calculations to create a simple reagent or compound inventory with amount debiting. Define "Initial amount" as a simple readout, "Amount debited" as a simple readout, and "Amount remaining" as a calculated readout with batch/protocol aggregation scope, so that you can keep track of amounts remaining for each batch separately. Assuming you have named your inventory protocol "Inventory", the calculation will be as follows:
[Inventory -> Initial amount (mg)] - sum( [ Inventory -> Amount debited (mg)]
Aggregate by batch and protocol