
The machine learning model extension is part of the CDD Visualization subscription, and requires activation by CDD support. Please contact us to learn how to try it for 30 days, or to subscribe to CDD Visualization.
Creating a model calls for two sets of molecules to train the model: the "good" molecules, and a previously screened training set. CDD Vault uses FCFP6 structural fingerprints to build a Bayesian statistical model. The model will then generate a score that can be used to rank compounds that have not been screened yet. The machine learning model is stored as a special type of protocol (category = Machine-Learning model), and provides a ROC plot so you can gauge its effectiveness. Each molecule receives a relative score, applicability number, and maximum similarity number.
The model will automatically score all compounds in the project you select while creating it. You can subsequently share it with other projects to score more molecules.
The models feature will be evolving significantly in the future. We'd love to hear what you want to be able to do with Machine-Learning models, to help guide that evolution.
If you prefer to watch a short demonstration on how to build a model, you can watch the following short video clip on our CDD Academy Youtube channel:
Machine-Learning
FCFP6 fingerprints
Bayesian Model
Baseline training set
"Good" molecules set
ROC curves
Score
Applicability
Maximum Similarity
Background
Machine-Learning
Machine-Learning is a computation technique of relating a molecular property to the underlying structure. Machine learning methods generally involves three broad steps: Firstly, the representation of a molecule in a way suitable for computerized treatment, often referred to as the choice of a descriptor (FCFP6 fingerprint). Secondly, the choice of the variable one attempts to model, often called the endpoint – which can be any molecular property that can be experimentally measured. Frequently used endpoints (since they are relevant in drug discovery practice) are solubility and logP as physicochemical properties, or bioactivity as a biological measurement variable (CDD protocol data). Thirdly, descriptors (input variables) and endpoints (output variables) need to be connected by means of one of a variety of available model generation methods (Bayesian model). From Bender, Molecular Similarity Primer
The following descriptors, and model are implemented by CDD. The end-point selection is performed by you, the experimenter, while building a model.
FCFP6 fingerprints
FCFP6 fingerprints are described in Rogers and Hahn 2010.
The chemical structure is examined for all subgraphs with a diameter of up to size 6 (i.e. start with a single node, and do 3 breadth-first iterations). Each of these graphs is assigned a hash code based on the properties of the atom, the bonds, and where applicable, chirality. These hash codes are put through several redundancy elimination steps, and eventually converted into a list of 32-bit integers. A druglike molecule typically has from dozens to hundreds of these unique hashcodes. Molecules that are structurally very similar tend to share a large number of these indices in common, and so are often compared using the Tanimoto coefficient. For ECFP-class fingerprints, the atom properties are somewhat literal (e.g. atomic number, charge, hydrogen count, etc.), whereas for the FCFP-class (“F” stands for functional) the atom characteristics are swapped out for properties that relate to ligand binding (e.g. hydrogen donor/acceptor, polarity, aromaticity, etc.) which means that different atoms often start with the same value (e.g. -OH and -NH might be considered the same).
Bayesian Model
A Naïve Bayesian model is optimized for sparse datasets, implemented as described in Xia et al. 2004.
The learned models are created with a straightforward learn-by-example paradigm: give it a set of hit compounds (the “good” samples), and the system learns to distinguish them from other baseline data. The learning process generates a large set of Boolean features from the input FCFP6 fingerprints, then collects the frequency of occurrence of each feature in the “good” subset and in all data samples. To apply the model to a particular compound, the features of the compound are generated, and a weight is calculated for each feature using a Laplacian-adjusted probability estimate. The model reports a score, which is calculated by normalizing the probability, taking the natural log and summing the results. This score is a relative predictor of the likelihood of that sample being from the “good” subset: the higher the score, the higher the likelihood. Once trained, the model can be applied to a set of compounds whose activity is unknown, and provides a score whose value gives a prediction of the likelihood that the molecule will be a hit in the modeled protocol.
Create a new model
Please contact us to activate the machine learning extension in your CDD Vault.
Once activated, you will notice a new "Build model" icon in the search results toolbar after a search is performed.

The model is built on two collections of molecules: The "good" molecules- the ones you'd like the model to consider as positives, and the "baseline" molecules, which are a large number of other types of molecules. Good molecules and baseline molecules do not necessarily mean active or inactive molecules in an assay, but the good set should include structures you wish to train the model to recognize amongst a large set of different kinds of structures represented by the baseline set.
"Training set baseline" molecules
Training set baseline molecules may come from the same protocol as the "good" set, a different protocol in your project, or a protocol in a public data-set. All of the molecules in the selected protocol will become part of the training baseline. This set should ideally contain many different kinds of molecules, including some of the "good" ones.
"Good" molecules
When you perform a search, all of the molecules in the search results will be considered part of the "good" set by default. For example, you can perform a search for hits of your secondary assay with IC50<10uM. You can also cherry-pick the good molecules from the search results by using the selection tool on the left of every structure: molecules are all selected by default, and you can click on the check-box to unselect it.
Fill in the model form

Your model will also need a name, and you will need to select a project where this model will be created.
Project selection is important, because once the model is created, all of the molecules in this project will automatically be scored. When the model is saved, you will see a green status bar with a hyperlinked model name. Click on the link to analyze results.
Analyze results
The model is created as a special kind of protocol in the project you selected. The new protocol is created automatically when you save a model. The protocol will not be editable, and will have a "Machine-Learning model" category, 3 readout definitions, and will have one automatically created run for scoring all molecules in the selected project.
ROC Curve
ROC curves are graphic representations of the relationship existing between the sensitivity and the specificity of a statistical test. It is generated by plotting the fraction of true positives (sensitivity) out of the total actual positives versus the fraction of false positives out of the total actual negatives (specificity). Gauge the effectiveness of the model by examining the curve: the best possible prediction method would yield a point in the upper left corner or coordinate (0,1) of the ROC space, representing 100% sensitivity (no false negatives) and 100% specificity (no false positives). The diagonal line represents random guessing. Points above the diagonal represent good classification results (better than random), points below the line poor results (worse than random).
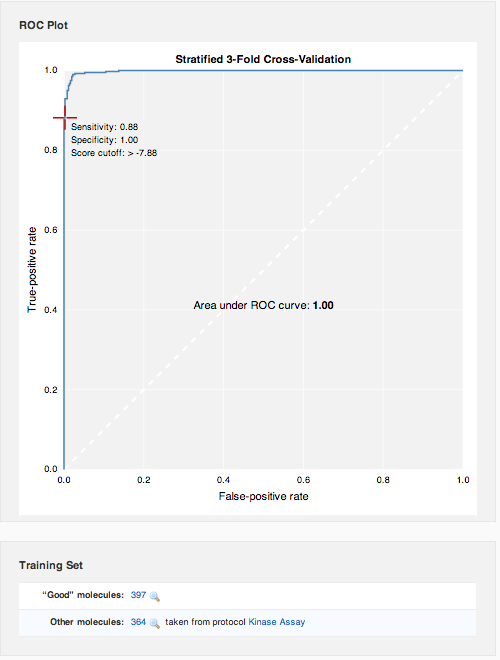
Right below the ROC curve, you can review the molecule sets that went into model creation.
Scoring of molecules
The protocol also contains readout definitions to store predicted ranking of molecules that have not been screened yet. Recall that as a model is created, it is assigned to a particular project. The model is applied to every molecule belonging to that project, and the results can be seen as a run of the machine learning model protocol. Click on the magnifying glass to view the results.

Score
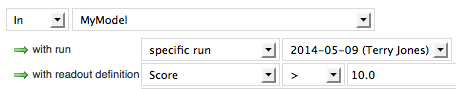
This score is a relative predictor of the likelihood of the molecule being a true positive according to the model: the higher the score, the higher the likelihood. To get an idea of the range of scores, sort the score column by clicking on the header in the search results table. Click again to sort from highest number to lowest. Now that you've got an idea of the range of possible scores, you can filter the molecules to show only high values.
Applicability
This is the fraction of structural features shared with the training set of molecules.
Maximum Similarity
Maximum Tanimoto/Jaccard similarity to any of the "good" molecules in the training set. This value is independent of the Bayesian model, and provides a way for you to perform a similarity search using all active compounds at once.
Apply model to untested compounds
To apply the model to other untested molecules in another project in your CDD Vault, all you need to do is share the protocol with that project. The underlying training set data used in the creation of the model will not be disclosed to the destination project if sharing permissions do not allow it. Nevertheless, all of the molecules belonging to the destination project will be automatically scored, and a new run with results will be added to the model protocol. The process may take several minutes, depending on the number of molecules to score, but when the yellow status bar at the top of the page disappears, all of the molecules will have been scored.
Look on the "Run data" tab of the model protocol, and click the magnifying glass icon as before.
The current version of the modeling functionality does not automatically score all new compounds which are added to a project after the model was last applied. To apply the model to new molecules, you will need to manually un-share and re-share the model with the current project. Here are the general steps:
- Make sure the model is shared with more than one project. You risk deleting the entire model if it's only shared with the one project you're interested in- you will delete this sharing in step 2.
- Once the model is shared with several projects, go to the Run data tab of the model protocol, and delete the old run for your project. To do so, click on the Run date, go to the Run Details tab, and "Delete this run" in the side-bar.
- Back on the Protocol Details form - un-share the model with your project of interest. Click "Manage project access" in the side-bar, and remove access from the project.
- Add back the access to this project you just removed in step 3. This will cause the model to run against all the compounds in the project, and a new run of the modeling protocol to be created from scratch.
The models feature will be evolving significantly in the future. We'd love to hear what you want to be able to do with machine learning models, to help guide that evolution.